computer science
OPENS DOORS
Publications by Shawn Martin
Please contact me for copies of these
articles if you want to read them and they are unavailable to you (contact). Also see
Google Scholar.
Book Chapters
-
H. K. Ho, L. Zhang, K. Ramamohanarao, and S. Martin (2013), "A Survey of Machine Learning Methods
for Secondary and Supersecondary Protein Structure Prediction," Protein Supersecondary Structure,
A. Kister, Ed., Humana Press (Publisher)
 In this chapter we provide a survey of protein secondary and supersecondary
structure prediction using methods from machine learning. Our focus is
on machine learning methods applicable to β-hairpin and β-sheet prediction,
but we also discuss methods for more general supersecondary structure
prediction. We provide background on the secondary and supersecondary
structures that we discuss, the features used to describe them, and the
basic theory behind the machine learning methods used. We survey the
machine learning methods available for secondary and supersecondary
structure prediction and compare them where possible.
In this chapter we provide a survey of protein secondary and supersecondary
structure prediction using methods from machine learning. Our focus is
on machine learning methods applicable to β-hairpin and β-sheet prediction,
but we also discuss methods for more general supersecondary structure
prediction. We provide background on the secondary and supersecondary
structures that we discuss, the features used to describe them, and the
basic theory behind the machine learning methods used. We survey the
machine learning methods available for secondary and supersecondary
structure prediction and compare them where possible.
-
M. Misra, S. Martin, and J.-L. Faulon (2011),
"Graphs: Flexible Representations of Molecular Structures and Biological Networks,"
Computational Approaches in Cheminformatics and Bioinformatics , R. Guha
and A. Bender, Eds., Wiley. & Sons. (Publisher)
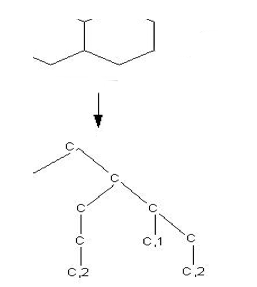 The past two decades have
seen a large accumulation of biological sequences and chemical
compounds in many publicly available databases. For a long time,
the two communities of bioinformatics and cheminformatics have
developed in parallel working largely with sequence data alone or
mainly in the chemical space, respectively. As the need to study
biological networks has increased, however, a concurrent need to
develop tools and algorithms capable of handling the combined sequence
and chemical space has arisen. We present here a graph-based
technique, named molecular signature, which is sufficiently adaptable
to permit combined description, for high-throughput analyses, of both
sequences and chemicals.
The past two decades have
seen a large accumulation of biological sequences and chemical
compounds in many publicly available databases. For a long time,
the two communities of bioinformatics and cheminformatics have
developed in parallel working largely with sequence data alone or
mainly in the chemical space, respectively. As the need to study
biological networks has increased, however, a concurrent need to
develop tools and algorithms capable of handling the combined sequence
and chemical space has arisen. We present here a graph-based
technique, named molecular signature, which is sufficiently adaptable
to permit combined description, for high-throughput analyses, of both
sequences and chemicals.
-
S. Martin (2010),
"Machine Learning based
Bioinformatics
Algorithms:
Application to Chemicals," Handbook
of Cheminformatics Algorithms, J.-L. Faulon and A. Bender, Eds.,
CRC Press. (publisher)
 In this chapter we present a targeted overview of clustering,
classification and regression algorithms. The target of our
overview is algorithms, which have been used in either bioinformatics
or chemoinformatics applications. In particular, we compare and
contrast the efforts in both fields.
In this chapter we present a targeted overview of clustering,
classification and regression algorithms. The target of our
overview is algorithms, which have been used in either bioinformatics
or chemoinformatics applications. In particular, we compare and
contrast the efforts in both fields.
-
S. Martin, W. M. Brown, and J.-L. Faulon (2008), "Predicting
Protein Interactions using Product Kernels," Advances in Biochemical
Engineering/Biotechnology: Protein-Protein Interactions, M.
Werther and H. Seitz, Eds., vol. 110, Springer-Verlag.
(publisher,
presentation)
 In this chapter, we provide a brief discussion of
the relative merits of different experimental and computational
methods available for identifying protein interactions. We
then focus on the application of our particular (computational) method
using Support Vector Machine product kernels. We describe
our method in detail and discuss the application of the method for
predicting protein–protein interactions, β-strand interactions, and
protein–chemical interactions.
In this chapter, we provide a brief discussion of
the relative merits of different experimental and computational
methods available for identifying protein interactions. We
then focus on the application of our particular (computational) method
using Support Vector Machine product kernels. We describe
our method in detail and discuss the application of the method for
predicting protein–protein interactions, β-strand interactions, and
protein–chemical interactions.
-
G. S. Davidson,
S. Martin, K.
Boyack,
B. N. Wylie, J. Martinez, A.
Aragon, M. Werner-Washburne, M. Mosquera-Caro, and C. L. Willman
(2007), "Robust
Methods in Microarray Analysis," Genomics
and Proteomics Engineering in Medicine and Biology
M. Akay, Ed., Wiley/IEEE.
(publisher)
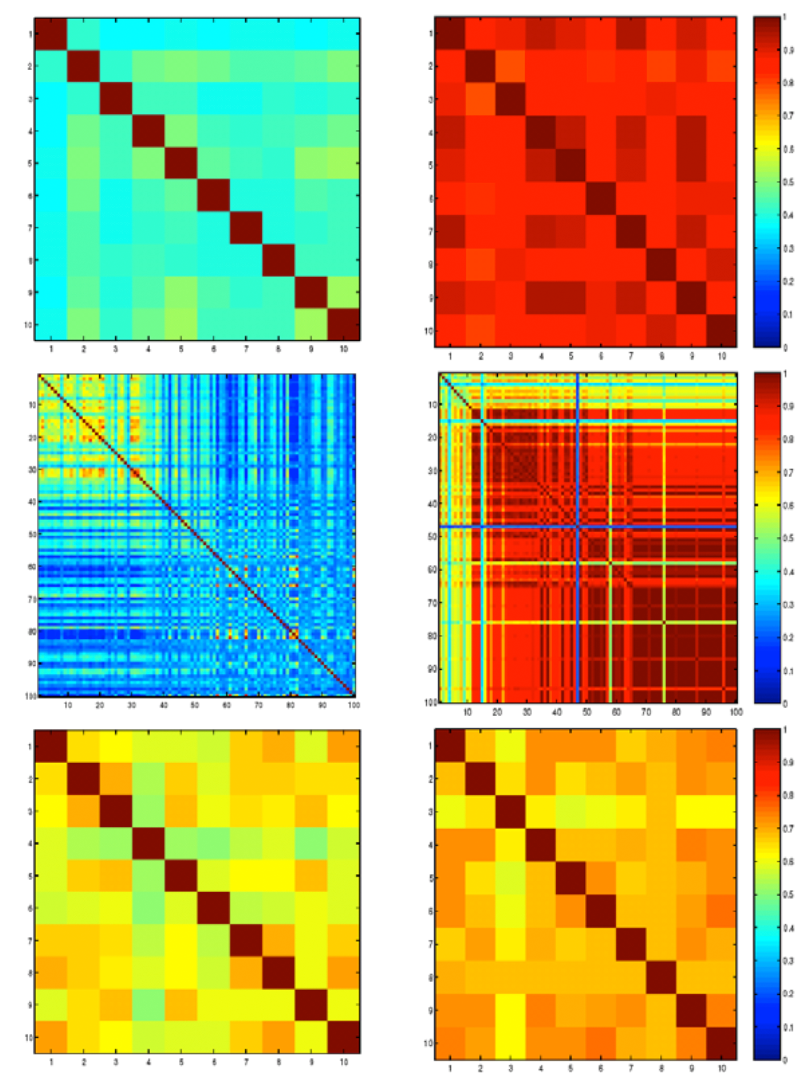 High throughput analysis techniques are required in
order to make good use of the genomic sequences that have recently
become available for many species, including humans.
Unfortunately, microarray data are also notoriously inaccurate, and it
is possible to spend far too much time contemplating the results of a
given microarray analysis method, only to arrive at a dead end.
In this chapter, we discuss several methods for microarray analysis we
have developed, which are meant to provide more accurate results and/or
quality assessments of the results obtained.
High throughput analysis techniques are required in
order to make good use of the genomic sequences that have recently
become available for many species, including humans.
Unfortunately, microarray data are also notoriously inaccurate, and it
is possible to spend far too much time contemplating the results of a
given microarray analysis method, only to arrive at a dead end.
In this chapter, we discuss several methods for microarray analysis we
have developed, which are meant to provide more accurate results and/or
quality assessments of the results obtained.
Journal Articles
-
S. Martin (2012), "Lattice Enumeration for Inverse Molecular Design Using the Signature
Descriptor," Journal of Chemical Information and Modeling, 52(7):1787-1797.
(journal, software)
 We describe an inverse quantitative structure–activity relationship (QSAR)
framework developed for the design of molecular structures with desired
properties. This framework uses chemical fragments encoded with a molecular
descriptor known as a signature. It solves a system of linear constrained
Diophantine equations to reorganize the fragments into novel molecular
structures. The method has been previously applied to problems in drug
and materials design but has inherent computational limitations due to the
necessity of solving the Diophantine constraints. We propose a new
approach to overcome these limitations using the Fincke–Pohst algorithm
for lattice enumeration. We benchmark the new approach against previous
results on LFA-1/ICAM-1 inhibitory peptides, linear homopolymers, and
hydrofluoroether foam blowing agents.
We describe an inverse quantitative structure–activity relationship (QSAR)
framework developed for the design of molecular structures with desired
properties. This framework uses chemical fragments encoded with a molecular
descriptor known as a signature. It solves a system of linear constrained
Diophantine equations to reorganize the fragments into novel molecular
structures. The method has been previously applied to problems in drug
and materials design but has inherent computational limitations due to the
necessity of solving the Diophantine constraints. We propose a new
approach to overcome these limitations using the Fincke–Pohst algorithm
for lattice enumeration. We benchmark the new approach against previous
results on LFA-1/ICAM-1 inhibitory peptides, linear homopolymers, and
hydrofluoroether foam blowing agents.
-
S. Martin and
J.-P. Watson (2011), "Non-Manifold
Surface Reconstruction from High Dimensional Point Cloud Data,"
Computational Geometry: Theory and
Applications, 44(8):427-441.
(journal,
software)
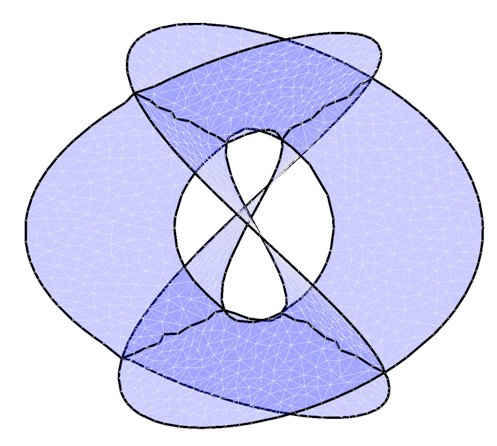 We describe an algorithm capable of reconstructing a
non-manifold surface embedded as a point cloud in a high dimensional
space. Our algorithm will work for non-orientable surfaces, and for
surfaces with certain types of self-intersection. The
self-intersections must be ordinary double curves and are fitted locally
by intersecting planes using a degenerate quadratic surface.
We describe an algorithm capable of reconstructing a
non-manifold surface embedded as a point cloud in a high dimensional
space. Our algorithm will work for non-orientable surfaces, and for
surfaces with certain types of self-intersection. The
self-intersections must be ordinary double curves and are fitted locally
by intersecting planes using a degenerate quadratic surface.
-
S. Martin, A.
Thompson, E. A. Coutsias, and J.-P. Watson (2010),
"Topology of Cyclo-Octane Energy
Landscape," Journal of
Chemical Physics
132:234115. (journal,
software, presentation)
 Understanding energy landscapes is a major challenge
in chemistry and biology. Although a wide variety of methods have been
invented and applied to this problem, very little is understood about
the actual mathematical structures underlying such landscapes. We have
discovered an example of an energy landscape which is nonmanifold,
demonstrating previously unknown mathematical complexity. The example
occurs in the energy landscape of cyclo-octane, which was found to have
the structure of a reducible algebraic variety, composed of the union
of a sphere and a Klein bottle, intersecting in two rings.
Understanding energy landscapes is a major challenge
in chemistry and biology. Although a wide variety of methods have been
invented and applied to this problem, very little is understood about
the actual mathematical structures underlying such landscapes. We have
discovered an example of an energy landscape which is nonmanifold,
demonstrating previously unknown mathematical complexity. The example
occurs in the energy landscape of cyclo-octane, which was found to have
the structure of a reducible algebraic variety, composed of the union
of a sphere and a Klein bottle, intersecting in two rings.
-
S. Martin, G.
Chandler, and M. S. Derzon (2008), "Simulation
of High Pressure Micro-Capillary 3He Counters," Journal of
Physics G: Nuclear and Particle Physics 35:115103. (journal)
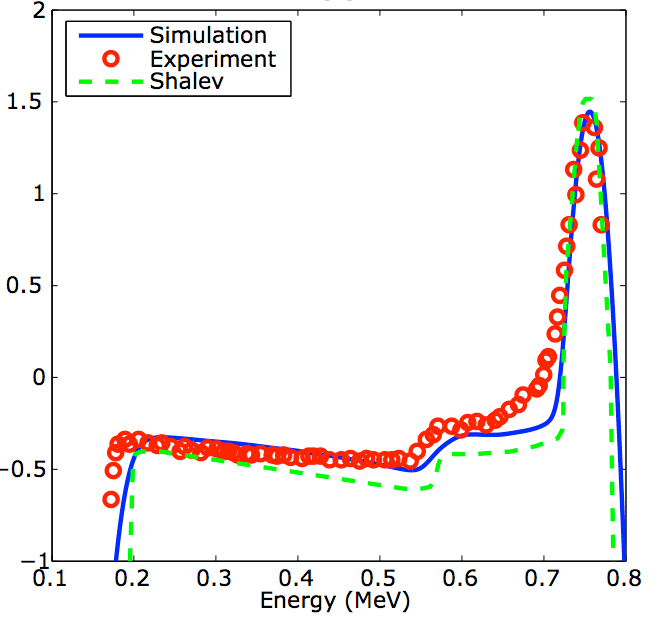 Low pressure (1-4 atm) cylindrical 3He counters are
widely used as neutron detectors. These detectors are relatively
large (1-2.5 cm diameter) and can be subject to noise induced by
microphonics. Meanwhile, new advancements in micro-fabrication
are enabling the manufacture of high pressure (over 3000 atm)
micro-capillaries (~100 micron diameter). Can these
micro-capillaries be used as accurate and high-efficiency 3He
counters? To investigate these questions, we have developed a
mathematical model/computer simulation.
Low pressure (1-4 atm) cylindrical 3He counters are
widely used as neutron detectors. These detectors are relatively
large (1-2.5 cm diameter) and can be subject to noise induced by
microphonics. Meanwhile, new advancements in micro-fabrication
are enabling the manufacture of high pressure (over 3000 atm)
micro-capillaries (~100 micron diameter). Can these
micro-capillaries be used as accurate and high-efficiency 3He
counters? To investigate these questions, we have developed a
mathematical model/computer simulation.
-
W. M. Brown, S.
Martin, S. N. Pollock, E. A. Coutsias, and J.-P. Watson
(2008), "Algorithmic Dimensionality
Reduction for Molecular
Structure Analysis," Journal
of
Chemical Physics 129(6):064118. (journal)
 Linear dimensionality reduction approaches have been
used to exploit the redundancy in a Cartesian coordinate representation
of molecular motion by producing low-dimensional representations of
molecular motion. Here, we investigate the effectiveness of
several
automated algorithms for nonlinear dimensionality reduction for
representation of trans,trans-1,2,4-trifluorocyclooctane conformation -
a molecule whose structure can be described on a 2-manifold in a
Cartesian coordinate phase space
Linear dimensionality reduction approaches have been
used to exploit the redundancy in a Cartesian coordinate representation
of molecular motion by producing low-dimensional representations of
molecular motion. Here, we investigate the effectiveness of
several
automated algorithms for nonlinear dimensionality reduction for
representation of trans,trans-1,2,4-trifluorocyclooctane conformation -
a molecule whose structure can be described on a 2-manifold in a
Cartesian coordinate phase space
-
W. M. Brown,
A. Sasson, D. R. Bellew, L. A. Hunsaker, S. Martin, A.
Leitao, L. M. Deck, D. L. Vander Jagt, and T. Oprea (2008),
"Efficient Calculation of Molecular
Properties from Simulation using
Kernel Molecular Dynamics," Journal
of Chemical Information and Modeling 48(8):1626-1637. (journal)
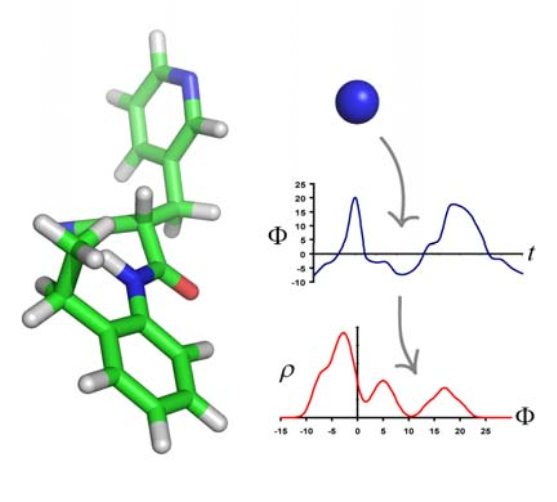 Understanding the relationship between chemical
structure and function is a ubiquitous problem in chemistry and
biology. Here, we present a novel approach that uses aspects of
simulation and informatics in order to formulate structure−property
relationships. We show how supervised learning can be utilized to
overcome the sampling problem in simulation approaches. Likewise,
we show how learning can be achieved based on molecular descriptions
that are rooted in the physics of dynamic intermolecular forces.
Understanding the relationship between chemical
structure and function is a ubiquitous problem in chemistry and
biology. Here, we present a novel approach that uses aspects of
simulation and informatics in order to formulate structure−property
relationships. We show how supervised learning can be utilized to
overcome the sampling problem in simulation approaches. Likewise,
we show how learning can be achieved based on molecular descriptions
that are rooted in the physics of dynamic intermolecular forces.
-
J.-L. Faulon, M.
Misra, S. Martin, K. Sale, and R. Sapra (2008),
"Genome Scale Enzyme-Metabolite and
Drug-Target Interaction
Predictions using the Signature Molecular Descriptor," Bioinformatics 24(2):225-233. (journal, pdf)
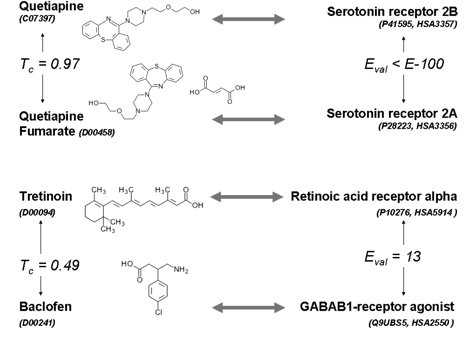 Identifying protein enzymatic or pharmacological activities
are important areas of research in biology and chemistry.
Biological and chemical databases are increasingly being populated
with linkages between protein sequences
and chemical structures. There is now sufficient information to
apply machine-learning techniques to predict interactions between
chemicals and proteins at a genome scale. Current
machine-learning techniques use as input
either protein sequences and structures or chemical information.
We propose here a method to infer
protein–chemical interactions using heterogeneous input consisting of
both protein sequence and chemical information.
Identifying protein enzymatic or pharmacological activities
are important areas of research in biology and chemistry.
Biological and chemical databases are increasingly being populated
with linkages between protein sequences
and chemical structures. There is now sufficient information to
apply machine-learning techniques to predict interactions between
chemicals and proteins at a genome scale. Current
machine-learning techniques use as input
either protein sequences and structures or chemical information.
We propose here a method to infer
protein–chemical interactions using heterogeneous input consisting of
both protein sequence and chemical information.
-
S. Martin, Z.
Zhang, A. Martino, and J.-L.
Faulon (2007), "Boolean
Dynamics of Genetic Regulatory Networks Inferred from Microarray Time
Series Data," Bioinformatics
23(7):866-874.
(journal,
pdf, supplement)
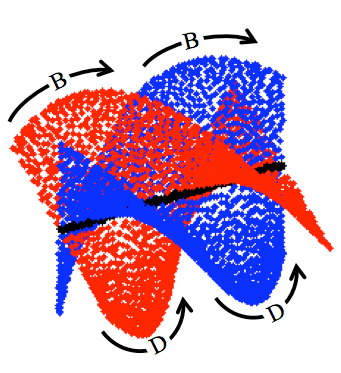
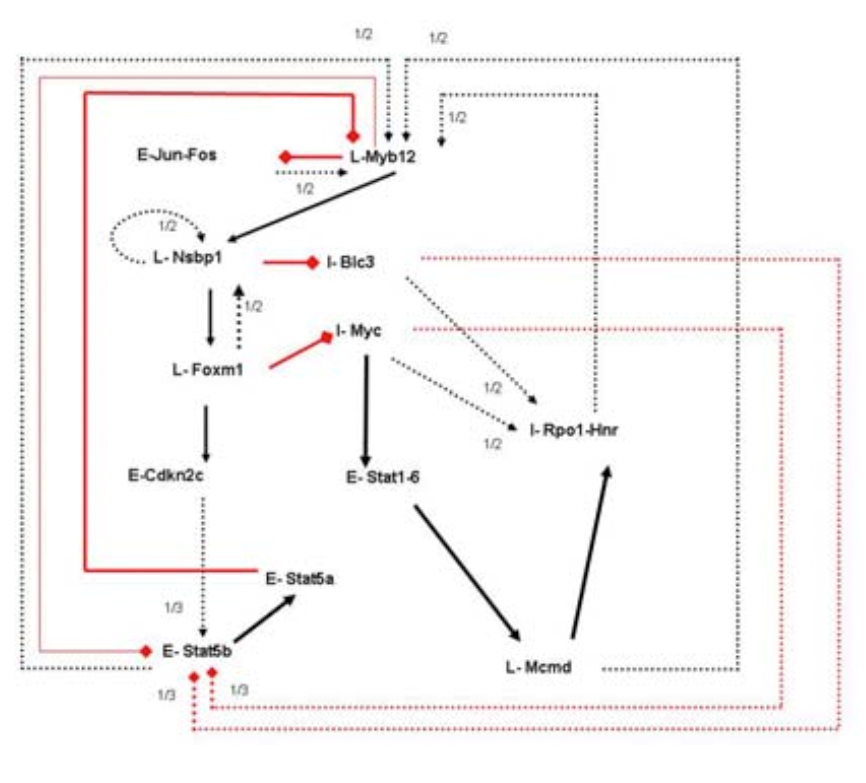 Methods available for the inference of genetic
regulatory networks strive to produce a single network, usually by
optimizing some quantity to fit the experimental observations. In
this paper we investigate the possibility that multiple networks can be
inferred, all resulting in similar dynamics. This idea is
motivated by theoretical work which suggests that biological networks
are robust and adaptable to change, and that the overall behavior of a
genetic regu-latory network might be captured in terms of dynamical
basins of attraction.
Methods available for the inference of genetic
regulatory networks strive to produce a single network, usually by
optimizing some quantity to fit the experimental observations. In
this paper we investigate the possibility that multiple networks can be
inferred, all resulting in similar dynamics. This idea is
motivated by theoretical work which suggests that biological networks
are robust and adaptable to change, and that the overall behavior of a
genetic regu-latory network might be captured in terms of dynamical
basins of attraction.
-
S. Martin, Z.
Mao, L. S. Chan, and S.
Rasheed (2007), "Inferring
Protein-Protein
Interaction Networks using Protein Complex Data," International Journal of Bioinformatics
Research and Applications 3(4):480-492. Expanded version
of BIOT 2006
conference paper with same authors. (journal)
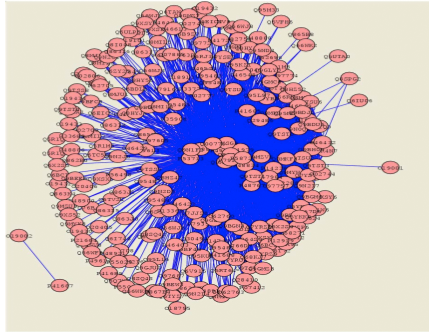 Present
day approaches for the determination
of protein-protein interaction networks are usually based on two hybrid
experimental measurements. Here we consider a computational
method that
uses another type of experimental data: instead of direct information
about protein-protein interactions, we consider data in the form of
protein complexes. We propose a method for using these complexes
to
provide predictions of protein-protein interactions. When applied
to a
dataset obtained from a cat melanoma cell line we find that we are able
to predict when a protein pair belongs to a complex with ∼96%
accuracy.
Present
day approaches for the determination
of protein-protein interaction networks are usually based on two hybrid
experimental measurements. Here we consider a computational
method that
uses another type of experimental data: instead of direct information
about protein-protein interactions, we consider data in the form of
protein complexes. We propose a method for using these complexes
to
provide predictions of protein-protein interactions. When applied
to a
dataset obtained from a cat melanoma cell line we find that we are able
to predict when a protein pair belongs to a complex with ∼96%
accuracy.
-
S. Martin, R. D.
Carr, and J.-L. Faulon (2006), "Random
Removal of
Edges from
Scale Free Graphs," Physica A
371(2):870-876. (journal)
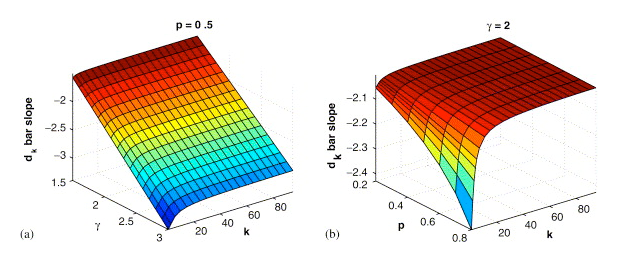 It
has been discovered that many naturally occurring networks (the
internet, the power grid of the western US, various biological
networks, etc.) satisfy a power-law degree distribution. Such scale-free
networks have many interesting properties, one of which is robustness
to random damage. This problem has been analyzed from the point
of view
of node deletion and connectedness. Recently, it has also been
considered from the point of view of node deletion and scale
preservation. In this paper we consider the problem from the
point of
view of edge deletion and scale preservation. In agreement with
the
work on node deletion and scale preservation, we show that a scale-free
graph should not be expected to remain scale free when edges are
removed at random.
It
has been discovered that many naturally occurring networks (the
internet, the power grid of the western US, various biological
networks, etc.) satisfy a power-law degree distribution. Such scale-free
networks have many interesting properties, one of which is robustness
to random damage. This problem has been analyzed from the point
of view
of node deletion and connectedness. Recently, it has also been
considered from the point of view of node deletion and scale
preservation. In this paper we consider the problem from the
point of
view of edge deletion and scale preservation. In agreement with
the
work on node deletion and scale preservation, we show that a scale-free
graph should not be expected to remain scale free when edges are
removed at random.
-
C. Wilson, G. S.
Davidson, S. Martin, E. Andries, J. Potter, R. Harvey, K. Ar, Y. Xu, K.
J. Kopecky, D. P. Ankerst, H. Gundacker, M. L. Slovak, M.
Mosquera-Caro, I-M. Chen, D. L. Stirewalt, M. Murphy, F. A. Shultz,
H. Kang, X. Wang, J. P. Radich, F. R. Appelbaum, S. R. Atlas, J.
Godwin, and C. L. Willman (2006), “Gene
Expression Profiling
of Adult Acute Myeloid Leukemia Identifies Novel Biologic Clusters for
Risk Classification and Outcome Prediction,”
Blood
108(2): 685-696. (journal,
pdf)
 To determine whether gene expression profiling could
improve risk classification and outcome prediction in older acute
myeloid leukemia (AML) patients, expression profiles were obtained in
pretreatment leukemic samples from 170 patients whose median age was 65
years. These expression profiles were analyzed using unsupervised
clustering methods were used to classify patients into 6 cluster groups
that varied significantly in rates of resistant disease. These
gene expression signatures provide insights into novel groups of AML
not predicted by traditional studies that impact prognosis and
potential therapy.
To determine whether gene expression profiling could
improve risk classification and outcome prediction in older acute
myeloid leukemia (AML) patients, expression profiles were obtained in
pretreatment leukemic samples from 170 patients whose median age was 65
years. These expression profiles were analyzed using unsupervised
clustering methods were used to classify patients into 6 cluster groups
that varied significantly in rates of resistant disease. These
gene expression signatures provide insights into novel groups of AML
not predicted by traditional studies that impact prognosis and
potential therapy.
-
W. M. Brown, S. Martin, Mark
D.
Rintoul, and J.-L. Faulon (2006), "Designing
Novel Polymers with
Targeted Properties using the Signature Molecular Descriptor," Journal of Chemical Information and
Modeling 46(2): 826-835. (journal)
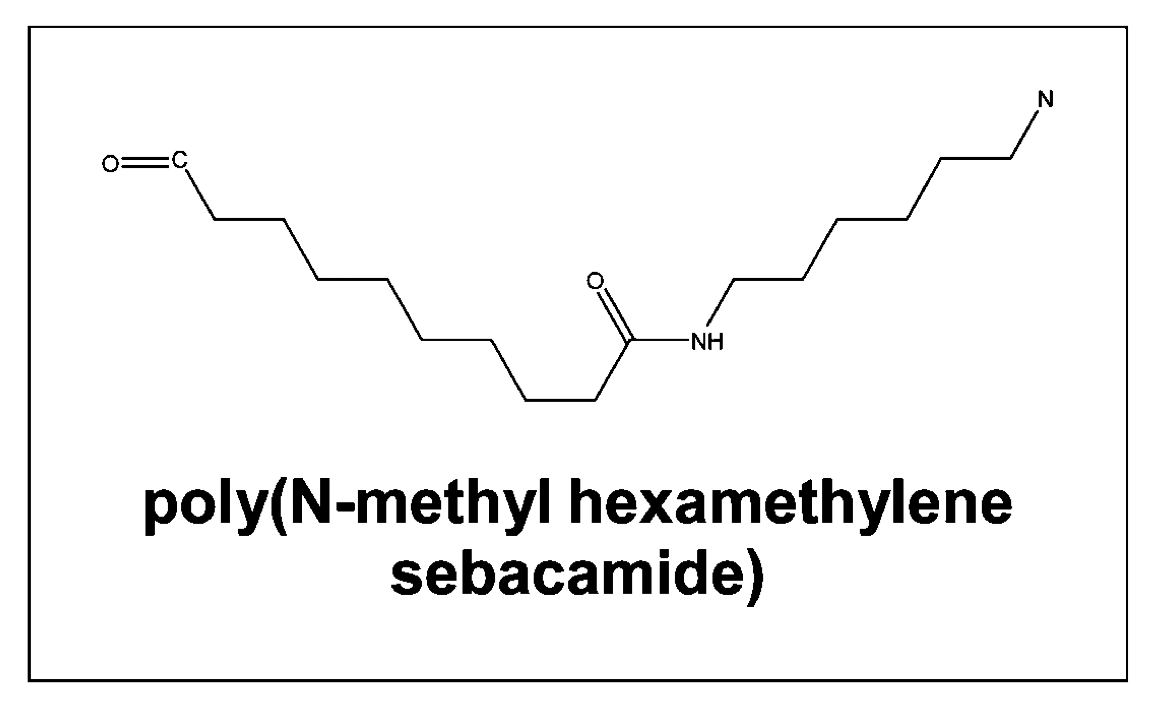 A method for solving the inverse quantitative
structure−property relationship (QSPR) problem is presented which
facilitates the design of novel polymers with targeted
properties. Here, we demonstrate the efficacy of the approach
using the targeted design of polymers exhibiting a desired glass
transition temperature, heat capacity, and density. We show how
the inverse problem can be solved to design poly(N-methyl hexamethylene
sebacamide) despite the fact that the polymer was used not used in the
training of this model.
A method for solving the inverse quantitative
structure−property relationship (QSPR) problem is presented which
facilitates the design of novel polymers with targeted
properties. Here, we demonstrate the efficacy of the approach
using the targeted design of polymers exhibiting a desired glass
transition temperature, heat capacity, and density. We show how
the inverse problem can be solved to design poly(N-methyl hexamethylene
sebacamide) despite the fact that the polymer was used not used in the
training of this model.
-
W. M. Brown,
S. Martin, J. Chabarek,
C. Strauss, and J.-L. Faulon (2006), "Prediction
of Beta-Strand
Packing Interactions using the Signature Product," Journal of Molecular Modeling
12(3):355-361. (journal,
poster)
 The prediction of β-sheet topology requires the
consideration of long-range interactions between β-strands that are not
necessarily consecutive in sequence. Since these interactions are
difficult to simulate using ab initio methods, we propose a
supplementary method able to assign β-sheet topology using only
sequence information. Our method is based on the signature
molecular descriptor, which has been used previously to predict
protein–protein interactions successfully, and to develop quantitative
structure–activity relationships for small organic drugs and peptide
inhibitors.
The prediction of β-sheet topology requires the
consideration of long-range interactions between β-strands that are not
necessarily consecutive in sequence. Since these interactions are
difficult to simulate using ab initio methods, we propose a
supplementary method able to assign β-sheet topology using only
sequence information. Our method is based on the signature
molecular descriptor, which has been used previously to predict
protein–protein interactions successfully, and to develop quantitative
structure–activity relationships for small organic drugs and peptide
inhibitors.
-
J.-L. Faulon, W. M. Brown, and S.
Martin
(2005), "Reverse
Engineering Chemical Structures from
Molecular Descriptors: How Many Solutions?," Journal of Computer
Aided Molecular Design 19(9-10):637-650. (journal)
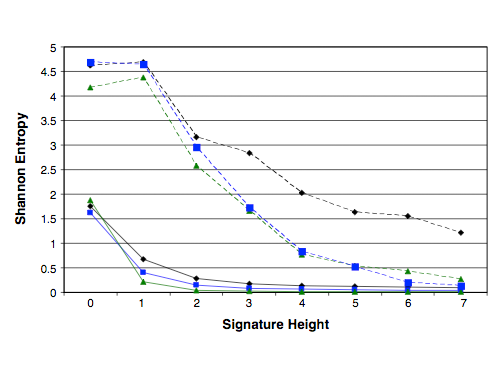 Physical,
chemical and biological properties are the ultimate
information of interest for chemical compounds. Molecular
descriptors
that map structural information to activities and properties are
obvious candidates for information sharing. In this paper, we
consider
the feasibility of using molecular descriptors to safely exchange
chemical information in such a way that the original chemical
structures cannot be reverse engineered.
Physical,
chemical and biological properties are the ultimate
information of interest for chemical compounds. Molecular
descriptors
that map structural information to activities and properties are
obvious candidates for information sharing. In this paper, we
consider
the feasibility of using molecular descriptors to safely exchange
chemical information in such a way that the original chemical
structures cannot be reverse engineered.
-
S. Martin, D. Roe, and J.-L. Faulon
(2005), "Predicting
Protein-Protein Interactions using Signature Products," Bioinformatics 21(2):218-226. (journal, pdf, software)
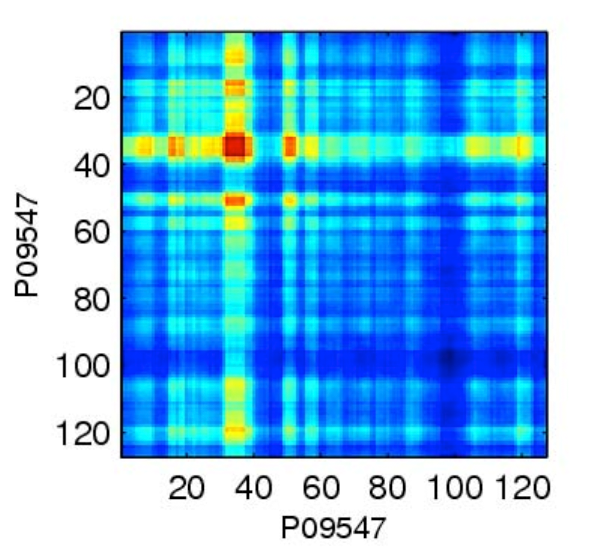 Proteome-wide prediction of protein–protein
interaction is a difficult and important problem in biology.
Although there have been recent advances in both experimental and
computational methods for predicting protein–protein interactions, we
are only beginning to see a confluence of these techniques. In
this paper, we describe a very general, high-throughput method for
predicting protein–protein interactions. Our method combines a
sequence-based description of proteins with experimental information
that can be gathered from any type of protein–protein interaction
screen.
Proteome-wide prediction of protein–protein
interaction is a difficult and important problem in biology.
Although there have been recent advances in both experimental and
computational methods for predicting protein–protein interactions, we
are only beginning to see a confluence of these techniques. In
this paper, we describe a very general, high-throughput method for
predicting protein–protein interactions. Our method combines a
sequence-based description of proteins with experimental information
that can be gathered from any type of protein–protein interaction
screen.
-
C. Churchwell, M. D. Rintoul, S.
Martin, D. P. Visco Jr., A. Kotu, R.
S. Larson, L. O. Sillerud, D. C. Brown, and J.-L. Faulon (2004), "The
Signature Molecular Descriptor 3. Inverse-Quantitative
Structure-Activity Relationship of ICAM-1 Inhibitory Peptides," Journal of Molecular
Graphics and Modeling
43(3):721-734. (journal)
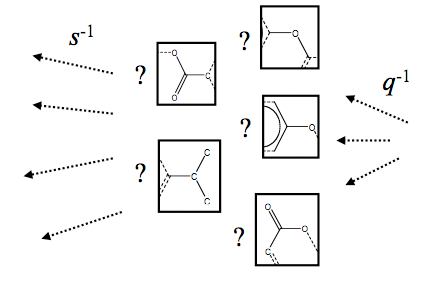 We present a
methodology for solving the inverse-quantitative
structure–activity relationship (QSAR) problem using the molecular
descriptor called signature. First, we create a QSAR equation
that correlates the occurrence
of a signature to the activity values using a stepwise multilinear
regression technique. Second, we construct constraint equations,
specifically the graphicality and consistency equations, which
facilitate the reconstruction of the solution compounds directly from
the signatures. Third, we solve the set of constraint equations,
which
are both linear and Diophantine in nature. Last, we reconstruct
and
enumerate the solution molecules and calculate their activity values
from the QSAR equation.
We present a
methodology for solving the inverse-quantitative
structure–activity relationship (QSAR) problem using the molecular
descriptor called signature. First, we create a QSAR equation
that correlates the occurrence
of a signature to the activity values using a stepwise multilinear
regression technique. Second, we construct constraint equations,
specifically the graphicality and consistency equations, which
facilitate the reconstruction of the solution compounds directly from
the signatures. Third, we solve the set of constraint equations,
which
are both linear and Diophantine in nature. Last, we reconstruct
and
enumerate the solution molecules and calculate their activity values
from the QSAR equation.
-
S. Martin, M. Kirby, and R. Miranda
(2000), "Symmetric
Veronese Classifiers with Application to Materials Design," Engineering
Applications of Artificial
Intelligence 13(5):513-520. (journal)
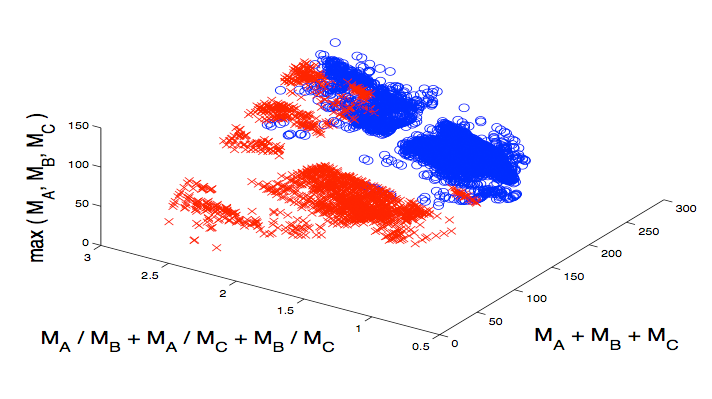 To solve the
materials classification problem, we propose a fast,
exhaustive approach. We propose to test every feature (chemical
property), every pair of features, every three features, etc., against
every classifier architecture from a certain group of classifiers known
as Support Vector Machines. This approach generalizes Pierre
Villars’
work to higher dimensions and more operations. We have
duplicated his result in identifying the Mendeleev Number as the single
best feature, and we have produced a new result for the case of two
features: namely, we have identified the Mendeleev number with the
valence electron number as the best combination of two features.
To solve the
materials classification problem, we propose a fast,
exhaustive approach. We propose to test every feature (chemical
property), every pair of features, every three features, etc., against
every classifier architecture from a certain group of classifiers known
as Support Vector Machines. This approach generalizes Pierre
Villars’
work to higher dimensions and more operations. We have
duplicated his result in identifying the Mendeleev Number as the single
best feature, and we have produced a new result for the case of two
features: namely, we have identified the Mendeleev number with the
valence electron number as the best combination of two features.
Letter to the Editor
-
S. Martin, M. P.
Mosquera-Caro, J. W.
Potter, G. S. Davidson, E. Andries, H. Kang, P. Helman, R. L. Veroff,
S. R. Atlas, M. Murphy, X. Wang, K. Ar, Y. Xu, I-M. Chen, F. A.
Schultz, C. S. Wilson, R. Harvey, E. Bedrick, J. Shuster, A. J.
Carroll, B. Camitta, and C. L. Willman (2007), "Gene
Expression Overlap affects Karyotype Prediction in Pediatric ALL,"
Leukemia
21:1341-1344. (journal)
 Treatment of acute
lymphoblastic leukemia (ALL) involves the assignment of patients to
risk groups based on cytogentic abnormalities. Here we report the
results of a gene expression experiment in which we have discovered
that the predictions of karyotype are insensitive, in that there are a
large number of false positive classifications among patients with
poorly defined cytogenetic abnormalities.
Treatment of acute
lymphoblastic leukemia (ALL) involves the assignment of patients to
risk groups based on cytogentic abnormalities. Here we report the
results of a gene expression experiment in which we have discovered
that the predictions of karyotype are insensitive, in that there are a
large number of false positive classifications among patients with
poorly defined cytogenetic abnormalities.
Conference Proceedings
-
S. Martin, V. Subramanya, and S. Mills
(2012), "Using Graph Layout
to Generalise Focus+Context Image Magnification
and Distortion," Image and Vision Computing New Zealand
(IVCNZ): 97-102. (proceedings, presentation)
 We present a novel framework for performing distortion-oriented focus+context image magnification. Our framework uses algorithms from graph drawing to manipulate the mesh underlying an image. Specifically, we apply a spectral graph layout algorithm to a weighted graph, where vertices in the graph correspond to pixels in the image, and edges connect directly adjacent vertices/pixels. By assigning appropriate weights to the edges, we can replicate the results of previous distortion-oriented approaches. In addition, we can perform image-aware distortion by using pixel values to influence the edge weights of our graph. We compare our approach to previous methods and demonstrate new results using image-based edge weighting schemes.
We present a novel framework for performing distortion-oriented focus+context image magnification. Our framework uses algorithms from graph drawing to manipulate the mesh underlying an image. Specifically, we apply a spectral graph layout algorithm to a weighted graph, where vertices in the graph correspond to pixels in the image, and edges connect directly adjacent vertices/pixels. By assigning appropriate weights to the edges, we can replicate the results of previous distortion-oriented approaches. In addition, we can perform image-aware distortion by using pixel values to influence the edge weights of our graph. We compare our approach to previous methods and demonstrate new results using image-based edge weighting schemes.
-
S. Martin, W. M.
Brown, R. Klavans,
and K. Boyack (2011), "OpenOrd: An
Open-Source Toolbox for Large Graph
Layout," Visualization and Data Analysis (VDA): 7868-06. (proceedings, software)
 We
document an open-source toolbox for drawing large-scale undirected
graphs. This toolbox is based on a previously implemented
closed-source algorithm known as VxOrd. Our toolbox, which we
call OpenOrd, extends the capabilities of VxOrd to large graph layout
by incorporating edge-cutting, a multi-level approach, average-link
clustering, and a parallel implementation. At each level,
vertices are grouped using force-directed layout and average-link
clustering. The clustered vertices are then re-drawn and the
process is repeated. When a suitable drawing of the coarsened
graph is obtained, the algorithm is reversed to obtain a drawing of the
original graph. This approach results in layouts of large graphs
which incorporate both local and global structure.
We
document an open-source toolbox for drawing large-scale undirected
graphs. This toolbox is based on a previously implemented
closed-source algorithm known as VxOrd. Our toolbox, which we
call OpenOrd, extends the capabilities of VxOrd to large graph layout
by incorporating edge-cutting, a multi-level approach, average-link
clustering, and a parallel implementation. At each level,
vertices are grouped using force-directed layout and average-link
clustering. The clustered vertices are then re-drawn and the
process is repeated. When a suitable drawing of the coarsened
graph is obtained, the algorithm is reversed to obtain a drawing of the
original graph. This approach results in layouts of large graphs
which incorporate both local and global structure.
-
S. Martin and S. McKenna, (2007),
"Predicting Building Contamination
using Machine Learning,"
International Conference on Machine Learning and Applications (ICMLA):
192-197. (proceedings,
presentation)
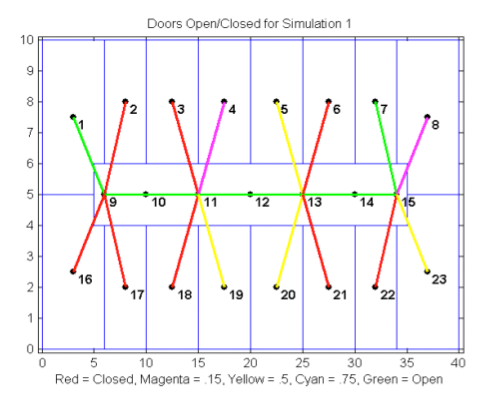 Potential events involving biological or chemical
contamination of buildings are of major concern in the area of homeland
security. Tools are needed to provide rapid, onsite predictions of
contaminant levels given only approximate measurements in limited
locations throughout a building. In principal, such tools could use
calculations based on physical process models to provide accurate
predictions. In practice, however, physical process models are too
complex and computationally costly to be used in a real-time scenario.
We investigate the feasibility of using machine learning
to provide easily computed but approximate models that would be
applicable in the field.
Potential events involving biological or chemical
contamination of buildings are of major concern in the area of homeland
security. Tools are needed to provide rapid, onsite predictions of
contaminant levels given only approximate measurements in limited
locations throughout a building. In principal, such tools could use
calculations based on physical process models to provide accurate
predictions. In practice, however, physical process models are too
complex and computationally costly to be used in a real-time scenario.
We investigate the feasibility of using machine learning
to provide easily computed but approximate models that would be
applicable in the field.
-
J. Joo, S.
Plimpton, S. Martin, L.
Swiler, and J.-L. Faulon (2007), "Sensitivity
Analysis
of a Computational Model of the
IKK-NF-kB-IkBa-A20 Signal Transduction Network," Annals of the New York Academy of Sciences
1115:221-239. (proceedings)
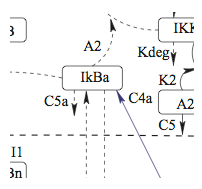 The NF-kB signaling network plays an important role
in many different compartments of the immune system during immune
activation. Using a computational model of the NF-kB signaling
network involving two negative regulators, IkBa and A20, we performed
sensitivity analyses with three different sampling methods and present
a ranking of the kinetic rate variables by the strength of their
influence on the NF-kB signaling response. We also present a
classification of temporal-response profiles of nuclear NF-kB
concentration into six clusters, which can be regrouped to three
biologically relevant clusters.
The NF-kB signaling network plays an important role
in many different compartments of the immune system during immune
activation. Using a computational model of the NF-kB signaling
network involving two negative regulators, IkBa and A20, we performed
sensitivity analyses with three different sampling methods and present
a ranking of the kinetic rate variables by the strength of their
influence on the NF-kB signaling response. We also present a
classification of temporal-response profiles of nuclear NF-kB
concentration into six clusters, which can be regrouped to three
biologically relevant clusters.
-
S. Martin (2006), "An
Approximate Version of Kernel PCA," Proceedings of the 5th
International Conference on Machine Learning and Applications
(ICMLA):239-244. (proceedings,
presentation,
poster)
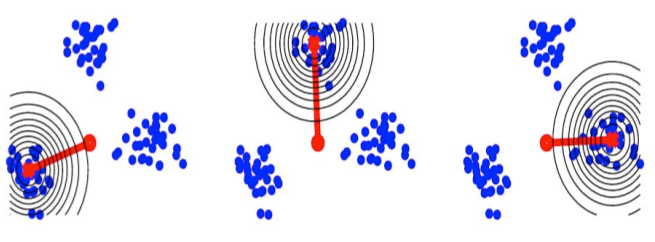 We propose an analog of kernel principal
component analysis (kernel
PCA). Our algorithm is based on an approximation of PCA which uses
Gram-Schmidt orthonormalization. We combine this approximation with
support vector machine kernels to obtain a nonlinear generalization of
PCA. By using our approximation to PCA we are able to provide a more
easily computed (in the case of many data points) and readily
interpretable version of kernel PCA.
We propose an analog of kernel principal
component analysis (kernel
PCA). Our algorithm is based on an approximation of PCA which uses
Gram-Schmidt orthonormalization. We combine this approximation with
support vector machine kernels to obtain a nonlinear generalization of
PCA. By using our approximation to PCA we are able to provide a more
easily computed (in the case of many data points) and readily
interpretable version of kernel PCA.
-
S. Martin, Z.
Mao, L. S. Chan, S.
Rasheed (2006), "Protein
Interactions Extrapolated from
Feline Protein Complexes," Proceedings of the 3rd Biotechnology
and
Bioinformatics Symposium
(BIOT):45-52. (pdf, presentation)
 The determination of protein-protein interaction networks is a
difficult problem in biology. Present day approaches to this
problem are usually based on two hybrid experimental measurements
coupled with refinement and extrapolation using computational
techniques. Here we consider a computational method for
similar refinement and extrapolation using experimental data from which
protein interactions can not be directly inferred.
The determination of protein-protein interaction networks is a
difficult problem in biology. Present day approaches to this
problem are usually based on two hybrid experimental measurements
coupled with refinement and extrapolation using computational
techniques. Here we consider a computational method for
similar refinement and extrapolation using experimental data from which
protein interactions can not be directly inferred.
-
S. Martin
(2006), "The
Numerical Stability of Kernel Methods," Proceedings of the 9th
International Symposium on Artificial Intelligence and Mathematics
(AIMATH):P01. (pdf, presentation)
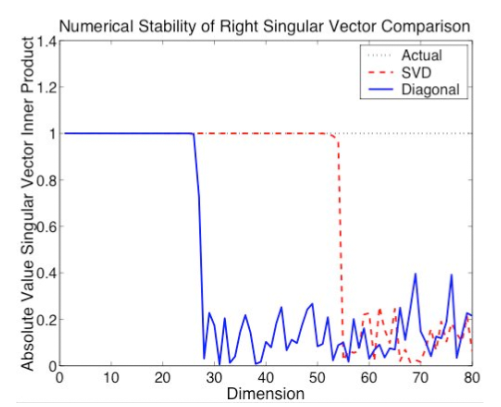 Kernel
methods use kernel functions to provide nonlinear versions of different
methods in machine learning and data mining, such as Principal
Component Analysis and Support Vector Machines. These kernel
functions require the calculation of some or all of the entries of a
matrix of the form XTX . The formation of this type of
matrix is known to result in potential numerical instability in the
case of least squares problems. How does the computation of the kernel
matrix impact the stability of kernel methods? We investigate
this question in detail in the case of kernel PCA and also provide some
analysis of kernel use in Support Vector Machines.
Kernel
methods use kernel functions to provide nonlinear versions of different
methods in machine learning and data mining, such as Principal
Component Analysis and Support Vector Machines. These kernel
functions require the calculation of some or all of the entries of a
matrix of the form XTX . The formation of this type of
matrix is known to result in potential numerical instability in the
case of least squares problems. How does the computation of the kernel
matrix impact the stability of kernel methods? We investigate
this question in detail in the case of kernel PCA and also provide some
analysis of kernel use in Support Vector Machines.
-
S. Martin
(2005),
"Training
Support Vector Machines using
Gilbert's Algorithm,"
Proceedings of the 5th IEEE
International Conference on Data Mining (ICDM):306-313. (proceedings, presentation, software)
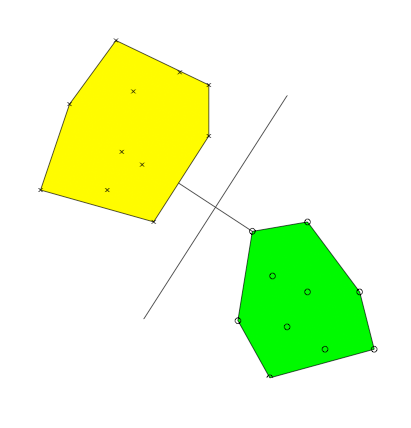 Support
vector machines are classifiers designed around the computation
of an optimal separating hyperplane. This hyperplane is typically
obtained by solving a constrained quadratic programming problem, but
may also be located by solving a nearest point problem. Gilbert's
algorithm can be used to solve this nearest point problem but is
unreasonably slow. In this paper we present a modified version of
Gilbert's algorithm for the fast computation of the support vector
machine hyperplane.
Support
vector machines are classifiers designed around the computation
of an optimal separating hyperplane. This hyperplane is typically
obtained by solving a constrained quadratic programming problem, but
may also be located by solving a nearest point problem. Gilbert's
algorithm can be used to solve this nearest point problem but is
unreasonably slow. In this paper we present a modified version of
Gilbert's algorithm for the fast computation of the support vector
machine hyperplane.
-
S. Martin and A.
Backer (2005),
"Estimating
Manifold Dimension by
Inversion Error," Proceedings of the 20th annual ACM Symposium
on
Applied Computing (SAC):22-26. (proceedings, presentation)
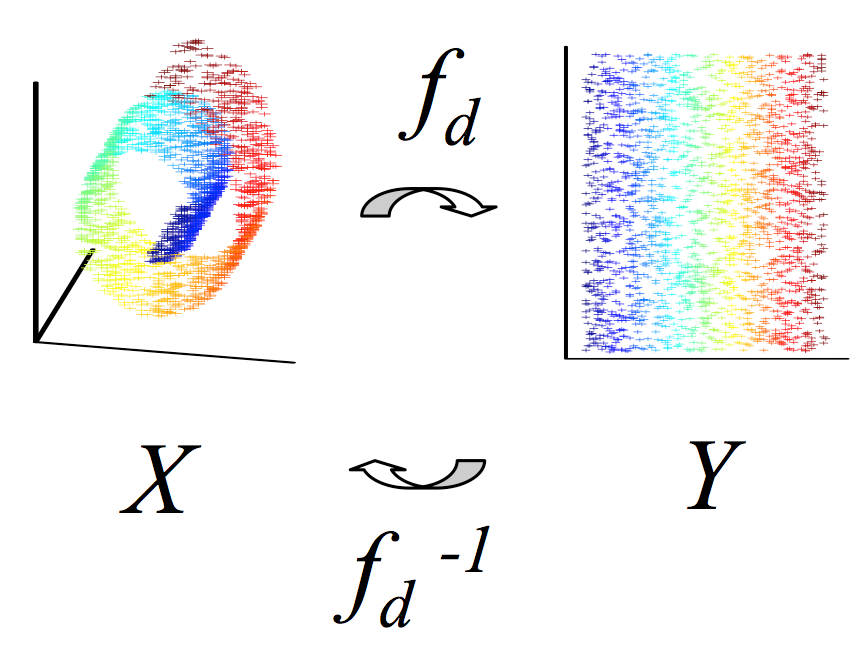 There has
been recent interest in
the application of a class of nonlinear dimensionality reduction
algorithms which assume that a dataset has been sampled from a
manifold. From this assumption, it follows that estimating the
dimension
of the manifold is the first step in analyzing an image dataset.
Once an estimate of the dimension is
obtained, it is used
as a parameter for the nonlinear dimensionality reduction
algorithm. In
this paper, we consider reversing this approach. Instead of
estimating
the dimension of the manifold in order to obtain a low dimensional
representation, we consider producing low dimensional representations
in order to estimate of the dimensionality of the manifold.
There has
been recent interest in
the application of a class of nonlinear dimensionality reduction
algorithms which assume that a dataset has been sampled from a
manifold. From this assumption, it follows that estimating the
dimension
of the manifold is the first step in analyzing an image dataset.
Once an estimate of the dimension is
obtained, it is used
as a parameter for the nonlinear dimensionality reduction
algorithm. In
this paper, we consider reversing this approach. Instead of
estimating
the dimension of the manifold in order to obtain a low dimensional
representation, we consider producing low dimensional representations
in order to estimate of the dimensionality of the manifold.
-
S. Martin, M. Kirby,
and R. Miranda
(2000), "Kernel/Feature
Selection for Support Vector Machines Applied to Materials Design,"
Proceedings of 9th IFAC Symposium on Artificial Intelligence in Real
Time
Control (AIRTC):29-34. (pdf)
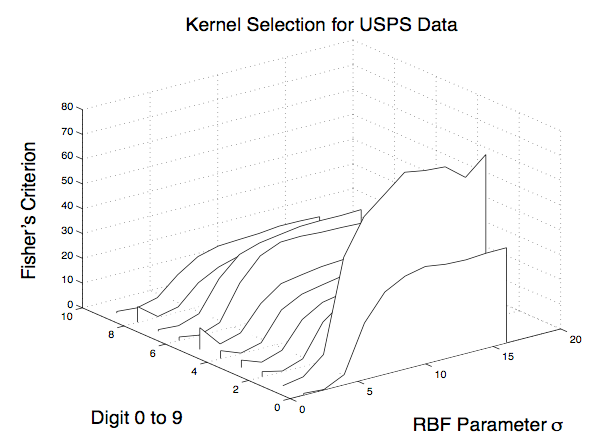 Support Vector Machines are classifiers with
architectures determined by kernel functions. In these
proceedings we propose a method for selecting the best SVM kernel for a
given classification problem. Our method searches for the best
kernel by remapping the data via a kernel variant of the classical
Gram-Schmidt orthonormalization procedure then using Fisher’s linear
discriminant on the remapped data.
Support Vector Machines are classifiers with
architectures determined by kernel functions. In these
proceedings we propose a method for selecting the best SVM kernel for a
given classification problem. Our method searches for the best
kernel by remapping the data via a kernel variant of the classical
Gram-Schmidt orthonormalization procedure then using Fisher’s linear
discriminant on the remapped data.
-
S. Martin, W. M.
Brown,
J.-L.
Faulon, D. Weis, D. Visco, and J. Kenneke (2005),
"Inverse
Design of Large Molecules using Linear Diophantine Equations,"
Proceedings of the 4th IEEE Computational Systems Bioinformatics
Workshops (CSBW):11-16. (proceedings, poster)
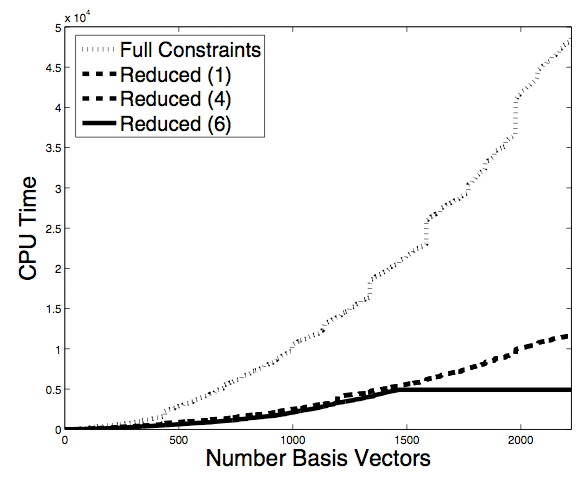 We have
previously developed a method for the inverse design of small
ligands. A
key step in our method involves computing the Hilbert basis of a system
of linear Diophantine equations. In our previous application, the
ligands considered were small peptide rings, so that the resulting
system of Diophantine equations was relatively small and easy to
solve.
When considering larger molecules, however, the Diophantine system is
larger and more difficult to solve. In this work we present a
method
for reducing the system of Diophantine equations before they are
solved, allowing the inverse design of larger compounds.
We have
previously developed a method for the inverse design of small
ligands. A
key step in our method involves computing the Hilbert basis of a system
of linear Diophantine equations. In our previous application, the
ligands considered were small peptide rings, so that the resulting
system of Diophantine equations was relatively small and easy to
solve.
When considering larger molecules, however, the Diophantine system is
larger and more difficult to solve. In this work we present a
method
for reducing the system of Diophantine equations before they are
solved, allowing the inverse design of larger compounds.
-
S. Martin, G. S.
Davidson, E.
E. May,
J.-L. Faulon, and M.
Werner-Washburne (2004), "Inferring
Genetic Networks from Microarray Data," Proceedings of the 3rd IEEE
Computational Systems
Bioinformatics (CSB):566-569. (proceedings, poster)
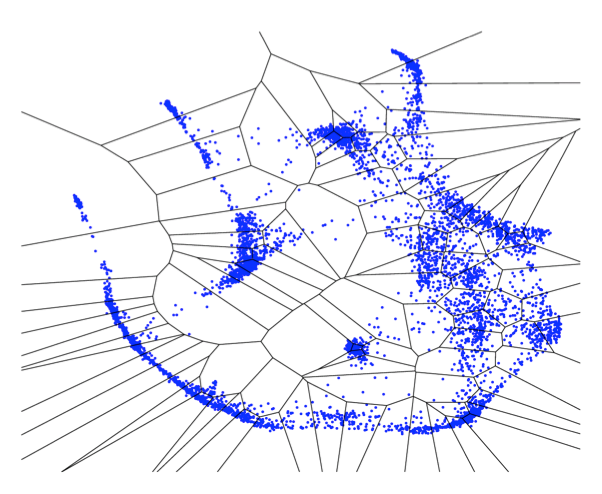 In
theory, it should be possible to infer realistic genetic networks
from time series microarray data. In practice, however, network
discovery has proved problematic. The three major challenges are
1)
inferring the network; 2) estimating the stability of the inferred
network; and 3) making the network visually accessible to the
user.
Here we describe a method, tested on publicly available time series
microarray data, which addresses these concerns.
In
theory, it should be possible to infer realistic genetic networks
from time series microarray data. In practice, however, network
discovery has proved problematic. The three major challenges are
1)
inferring the network; 2) estimating the stability of the inferred
network; and 3) making the network visually accessible to the
user.
Here we describe a method, tested on publicly available time series
microarray data, which addresses these concerns.
-
J.-L. Faulon, S. Martin, and R. D.
Carr
(2004), "Dynamical
Robustness in Gene Regulatory Networks," Proceedings of the 3rd IEEE
Computational Systems
Bioinformatics (CSB):626-627. (proceedings, pdf, poster)
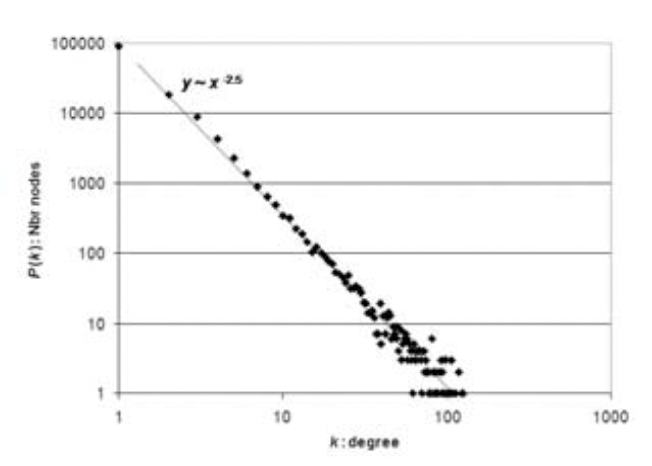 We investigate the robustness of biological
networks, emphasizing gene regulatory networks. We define the
robustness of a dynamical network as the magnitude of perturbation in
terms of rates and concentrations that will not change the steady state
dynamics of the network. We find the number of dynamical networks
versus their dynamical robustness follows a power law.
We investigate the robustness of biological
networks, emphasizing gene regulatory networks. We define the
robustness of a dynamical network as the magnitude of perturbation in
terms of rates and concentrations that will not change the steady state
dynamics of the network. We find the number of dynamical networks
versus their dynamical robustness follows a power law.
-
S.
Martin
(2001), Techniques
in Support Vector Classification,
Ph. D. Dissertation, Colorado State University. (pdf)
 Here we
consider three problems in Support Vector Classification: feature
selection, kernel selection, and training. Feature selection is
done using Fisher's discriminant adapted to SVMs. Kernel
selection is done using a kernel version of Gram-Schmidt
orthonormalization, and training is done using a geometrical
interpretation of the quadratic optimization program normally used to
solve for the SVM.
Here we
consider three problems in Support Vector Classification: feature
selection, kernel selection, and training. Feature selection is
done using Fisher's discriminant adapted to SVMs. Kernel
selection is done using a kernel version of Gram-Schmidt
orthonormalization, and training is done using a geometrical
interpretation of the quadratic optimization program normally used to
solve for the SVM.
-
S.
Martin
(1997), "Concerning the
Quadratic Relations which
define the Grassman Manifold," M.S. Paper, Colorado State
University. (pdf)
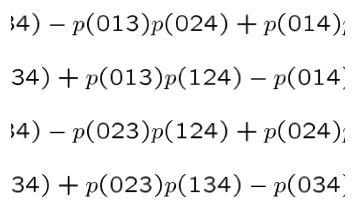 The Plucker
embedding gives a bijective correspondence between the d-planes of a
projective space Pn and the points of the Grassman Manifold
in a higher dimensional space PN. The Grassman Manifold can be
defined as the set of points in PN whose homogeneous coordinates satisfy
certain quadratic relations, those relations being generated by
sequences in {0,...,n}. Here we present a minimal set of
generating sequences for the quadratic relations and subsequently
investigate the linear independence of said relations.
The Plucker
embedding gives a bijective correspondence between the d-planes of a
projective space Pn and the points of the Grassman Manifold
in a higher dimensional space PN. The Grassman Manifold can be
defined as the set of points in PN whose homogeneous coordinates satisfy
certain quadratic relations, those relations being generated by
sequences in {0,...,n}. Here we present a minimal set of
generating sequences for the quadratic relations and subsequently
investigate the linear independence of said relations.
